
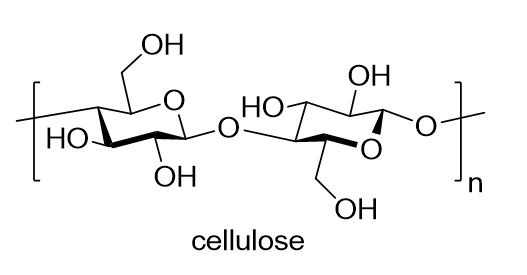
Les polymères (du grec polus, plusieurs et meros, partie) sont des macromolécules constituées de motifs qui se répètent. Leurs poids moléculaires sont supérieurs à 2000 g/mol. Les protéines, l’ADN, les polysaccharides, la cellulose ou l’amidon sont des polymères fabriqués par la nature. La cellulose (dessin ci-dessous) est un polymère constitué de molécules de D-glucose liées par des liaisons covalentes (des fonctions éthers). Ce glucide linéaire est le principal constituant de la paroi des cellules végétales. La cellulose constitue la matière organique la plus abondante sur la Terre (plus de 50% de la biomasse). La quantité synthétisée par les végétaux est estimée à 50-100 milliards de tonnes par an.
On peut séparer les polymères en deux grandes familles : les polymères naturels et les polymères synthétiques (qui sont synthétisés par l’humain). En fait, nous n’avons fait que copier la nature : d’abord en modifiant chimiquement les polymères naturels pour obtenir de nouvelles propriétés (le papier à partir du bois par exemple) puis en inventant de nouveaux procédés de polymérisation pour créer des matériaux aux propriétés innovantes et adaptées à nos besoins (les oreillers à mémoire de forme par exemple). Comment en sommes-nous arrivés là?
L’histoire des polymères
L’humain utilise depuis des siècles les polymères naturels pour en faire des outils, des objets ou des tissus (coton, soie). Le papier, par exemple, est fabriqué à partir de cellulose. Mais c’est seulement au début du XIXème siècle que les chimistes se sont intéressés à la modification chimique des polymères naturels pour faire de nouveaux matériaux.
1 – Le latex et le caoutchouc naturel
Thomas Hancock (1820) et Charles Goodyear (1839) seront les premiers à utiliser le latex comme matière première pour la fabrication du caoutchouc. Le latex qui coule de l’arbre (cf encadré) est constitué de longues chaînes de poly(cis-1,4-isoprène) (structure ci-dessous).
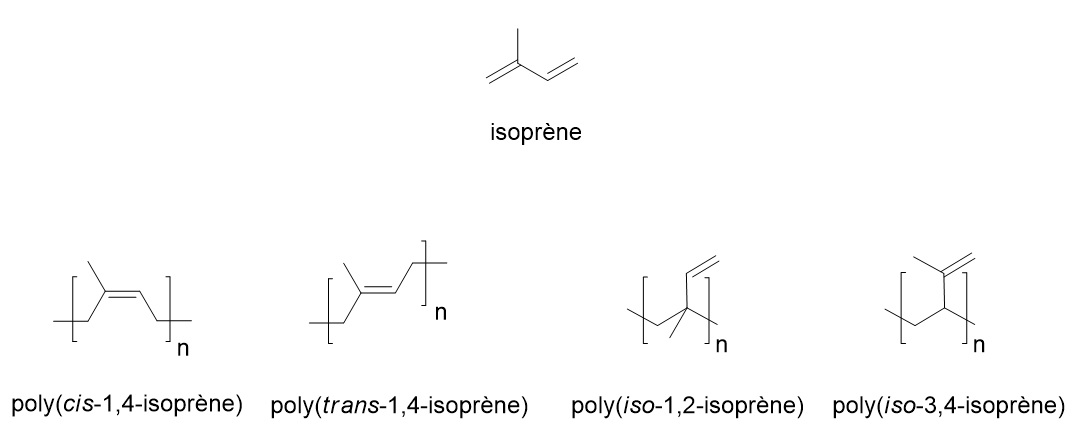
Ces longues chaînes forment en coulant de l’hévéa une huile visqueuse, collante, blanchâtre, flexible et résistante à température ambiante : on parle d’un polymère amorphe. On n’est encore loin d’un matériau intéressant. Hancock découvrira que la mastication favorise le moulage, tandis que Goodyear sera à l’origine de la découverte de la vulcanisation.

Le latex, issu de l’hévéa est différent de la sève. Alors que la sève distribue l’eau, les sels minéraux et les sucres, le latex est impliqué dans les mécanismes de défense de l’arbre. Il circule dans les canaux laticifères. Comme la résine, il suinte lors d’une éventuelle blessure de la plante et forme en séchant une barrière protectrice. Le latex se récolte par saignée. Un arbre peut produire du latex à partir de 5 ans et pendant 30 ans environ. La Thaïlande est le premier pays producteur. Le cultivateur dépose une tasse sous la saignée et le latex coule pendant quelques heures puis l’encoche se bouche. La récolte peut se faire sous forme liquide (récolte en latex) ou bien sous forme solide (récolte en coagulum). En cas de récolte sous forme liquide, on peut rajouter un peu d’ammoniaque pour éviter la coagulation ou bien de l’acide formique pour le faire coaguler. Les coagulum sont lavés, déchiquetés, rééduqués en granulés puis chauffés à 120°C. Le caoutchouc sans soufre est cassant à froid et poisseux à chaud.
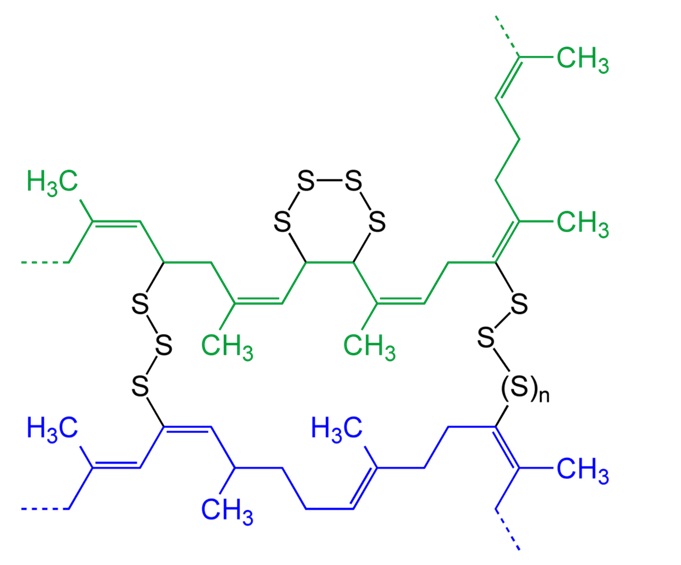
En 1844, Charles Goodyear dépose le premier brevet sur la modification d’un polymère : la vulcanisation. Il décrit que l’addition d’une certaine quantité de soufre à haute température permet l’obtention d’un nouveau matériau aux propriétés élastiques et non collante. Il aura inventé le caoutchouc naturel que l’on connait. Lorsque ce processus est prolongé de façon extrême et en présence d’une grande quantité de soufre (entre 20 et 40%), il se forme un solide noir, dur et cassant. Puisque ce matériau est noir comme l’ébène, il sera nommé ébonite ou vulcanite (cf encadré).

Lorsque du soufre est ajouté à chaud au latex, il se forme des structures cycliques intramoléculaires : les chaines linéaires de polymères se lient entre elles par des ponts (dits ponts disulfures). On parle de réticulation des chaînes linéaires. La vulcanisation est en fait la réaction du soufre sur les double liaisons des chaînes poly(isoprène). Plus la quantité de soufre ajoutée est grande et plus le matériau possède de réticulations. Le réseau 3D devient très dense et le matériau plus compact.
L’ébonite, appelée aussi vulcanite, inventé par Charles Goodyear en 1839 mais breveté en 1851 par son frère Nelson, est un matériau noir, dur et cassant. Il peut être additionné de charges : craie, magnésie, oxydes métalliques ou baryte. Ses applications seront nombreuses : boules de bowling, embouts de pipes ou d’instruments de musique, corps de stylos à plume. L’ébonite peut être polie et tournée comme du bois mais reste néanmoins cassante et fragile.
Après plusieurs années, Charles Goodyear va multiplier les inventions (canots de sauvetage, ressorts, vêtements) mais de nombreux contrats lui seront défavorables. Hutchinson rachètera le brevet de Goodyear et créera la marque « A l’Aigle » en 1853 en installant sa première usine de fabrication de vêtements en caoutchouc en France.
2 – La nitrocellulose : fulmicoton et collodion
Malgré les interdictions de sa femme, Christian Friedrich Schönbein expérimentait volontiers dans la cuisine familiale. En 1845, c’est en essuyant des tâches d’acide nitrique et d’acide sulfurique avec un torchon en coton qu’il fit sécher au-dessus du poêle qu’il découvrit le fulmicoton. Le torchon s’enflamma spontanément. Il venait de découvrir une alternative à la poudre à canon qui dégageait une fumée noire et compacte, qui salissait les artilleurs et encrassait les canons. La nitrocellulose, explosif fulminant, donnait la clef d’une poudre sans fumée. Les tentatives de production industrielle furent compliquées car les usines explosaient les unes après les autres. Il fallut attendre 1891 pour que Dewar et Abel parviennent à stabiliser le fulmicoton. Pour synthétiser de la nitrocellulose, on traite de la cellulose (du coton) avec de l’acide nitrique (HNO3) et de l’acide sulfurique (H2SO4). Les ions nitronium (NO2+) ainsi formés réagissent avec les fonctions alcools de la cellulose.
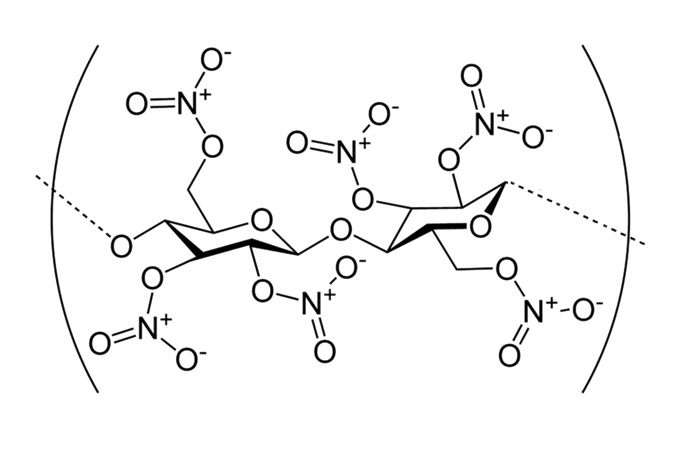
On obtient alors des mononitrates, dinitrates et trinitrates. Alors que le mononitrate est seulement inflammable et trouve des applications en lutherie, comme vernis à ongles ou comme collodion médicinal (pansement liquide), les dinitrates et trinitrates sont explosifs, solubles dans l’acétone et doivent être stockés dans l’eau pour être stabilisés. La nitrocellulose fut également utilisée comme support de films photographiques et cinématographiques. La technique photographique du collodion humide est un procédé attribué à l’anglais Frederick Scott Archer en 1851. Il remplaça l’albumine qui servait de medium pour fixer les sels d’argent sur des plaques de verre.

3 – Le celluloïd : la première matière plastique
Le celluloïd est la toute première matière plastique artificielle créé en 1870 par les frères Hyatt. Il s’agit de nitrate de cellulose mélangé à du camphre (qui sert de plastifiant). Pendant la guerre de Sécession, l’importation de l’ivoire d’éléphant qui est utilisée dans la fabrication des boules de billard est interdite. La société Phellan & Collender lance un concours d’inventeur pour remplacer l’ivoire par une nouvelle matière plastique aux propriétés similaires. John Wisley Hyatt recouvre une boule de billard avec du collodion dans le but de remporter le concours, mais le matériau est trop fragile. C’est en 1870, que lui et son frère ajoutent alors du camphre. Le celluloïd a été utilisé pour la fabrication des balles de tennis de table jusqu’en 2014, lorsque le matériau a été remplacé par un composite.

Le celluloïd était aussi utilisé dans la production de films cinématographiques : les films flamme. Par la suite, il a été remplacé par le triacétate de cellulose beaucoup moins inflammable.
4 – La viscose ou soie artificielle
Hilaire de Chardonnet et Auguste Delubac inventent la viscose en 1884 en mélangeant collodion et cellulose. Ils obtiennent une matière semblable à la soie : fine et brillante, mais beaucoup plus économique. Aujourd’hui la viscose est synthétisée industriellement à partir de cellulose et de disulfure de carbone (CS2).
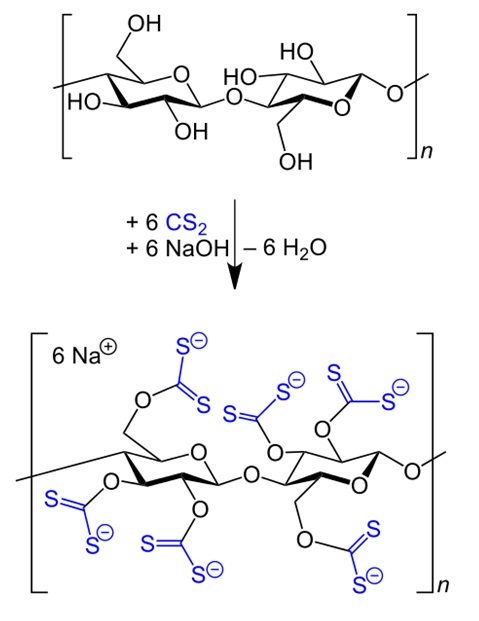
5 – Les premiers polymères synthétiques
La bakélite, inventé par Leo Baekeland est le premier polymère entièrement synthétisé par l’humain lors d’une réaction de polymérisation. La polycondensation du phénol et du formaldéhyde produit de l’eau, de la chaleur et une résine thermodurcissable phénoplaste.
On crée alors un réseau 3D très compact. Par la suite, différents matériaux seront étudiés par les chimistes : les élastomères à base d’isoprène (1914), les polyesters et les polyamides (1931 Wallace Carothers), le plexiglass, le polystyrène (1930) et le polychlorure de vinyle (1938).

La production de matières plastiques aujourd’hui
La production de matières plastiques s’élève à 300 millions de tonnes par an et cette production ne cesse de croître depuis les années 1950. L’invention du plastique moderne sonna comme une révolution. Il a permis de faciliter le quotidien des revenus modestes (notamment celui des femmes au foyer) : emballages jetables ou boîtes en plastiques (les fameux Tupperware), sacs de supermarché, électroménager et même mobilier !

L’époque où il était le bienvenu dans tous les foyers de France fut un pic de notre civilisation. La France jouissait alors d’une forte croissance économique, du plein emploi, d’une industrie surpuissante. Dans ses « Mythologies », essai paru en 1957, Roland Barthes célébrait cette « matière miraculeuse », cette « substance alchimique » qui était, dans une certaine mesure, le contraire du poujadisme : un arrachement à la tradition, à la terre, au bon sens. Le plastique, lui, ne ment pas ; il se donne pour ce qu’il est : un pur artifice façonné par l’homme, un produit de la chimie qui ne doit rien à la nature. Résolument moderne, nous disait Barthes ! Et pour cause, les plastiques issus de l’industrie chimique et plus directement de la raffinerie pétrolière ont de multiples qualités : bon marché, hydrophobes, inertes biologiquement, de faible densité, déformables, d’une grande résistance mécanique et chimique. Ils sont des matières premières de choix pour une utilisation à usage unique : couverts jetables, seringues, cotons tiges, sacs plastiques… Aux yeux du consommateur, le plastique n’a absolument aucune valeur. L’éventail des polymères de commodité est assez restreint comme le montre la répartition ci-dessous : polytéréphtalate d’éthylène pour les boissons (PETE ou PET), polyéthylène haute densité des sacs plastiques ou bidons de lessive (HDPE), polychlorure de vinyle des canalisations (PVC ou V), polyéthylène basse densité des emballages souples, polypropylène des boîtes alimentaires (PP), polystyrène des gobelets et des cintres (PS) et quelques autres : polyuréthanes, polyacryliques, résines thermodurcissables, etc.
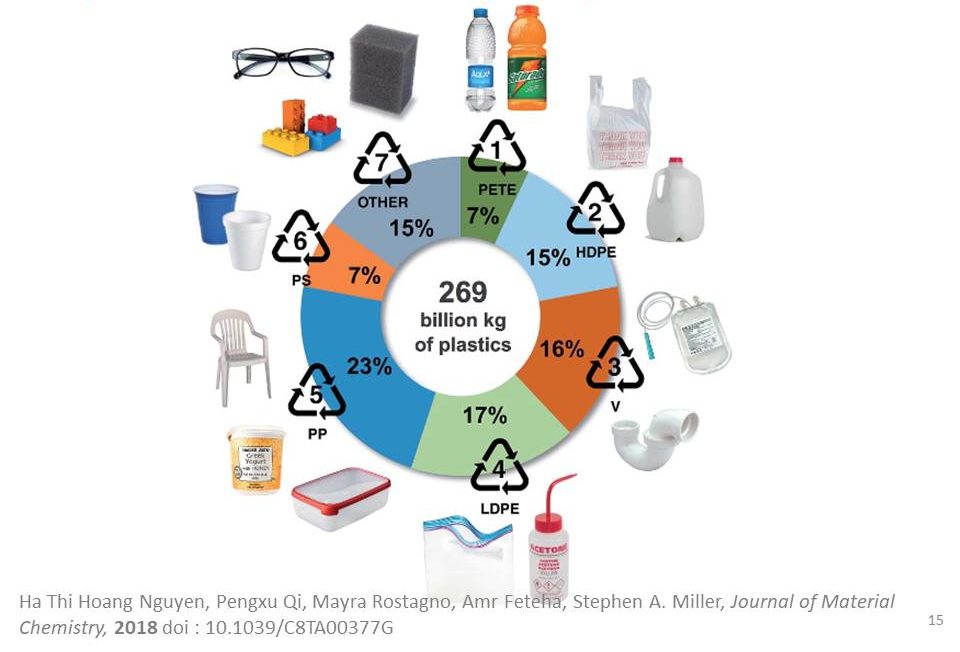
Le polypropylène (PP), le polyéthylène (PE), le polychlorure de vinyle (PVC) et le polystyrène (PS) représentent à eux seuls 80% de la production mondiale. Ils sont tous les quatre constitués de liaisons carbone-carbone apolaires difficiles à rompre qui les rend résistants aux attaques chimiques et biologiques.
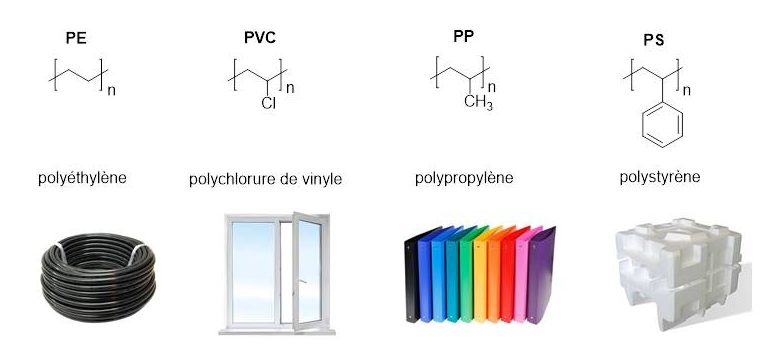
1 – Classification et caractéristiques
Les polymères sont synthétisés à façon en fonction des applications désirées. Il est possible de faire varier la nature des monomères utilisés pour la polymérisation, mais aussi la longueur des chaînes polymériques, les propriétés thermiques, les architectures et l’enchaînement des motifs de répétition. Et tout cela en faisant varier la réaction de polymérisation utilisée, les réactifs et les solvants mais aussi le traitement thermique après synthèse.
Un polymère dont les chaînes sont constituées de la répétition d’un seul monomère est un homopolymère, alors qu’un copolymère contient plusieurs monomères différents. Il existe différentes familles de copolymères en fonction de l’enchaînement de ces différents monomères : copolymères alternés, copolymères bloc, copolymères gradients et copolymères statistiques. Un copolymère bloc constitué de deux monomères différents A et B formera des chaînes : AAAAAAAAA-BBBBBBBBB. Un copolymère de ce type dont un bloc sera hydrophile et l’autre hydrophobe pourra par exemple être utilisé dans les crèmes cosmétiques pour stabiliser une émulsion huile dans eau (on parlera alors de surfactant polymérique). Le polytéréphtalate d’éthylène (PET) de nos bouteilles d’eau est un copolymère alterné issu de la polycondensation de l’éthylène glycol et de l’acide téréphtalique (dessin ci-dessous).
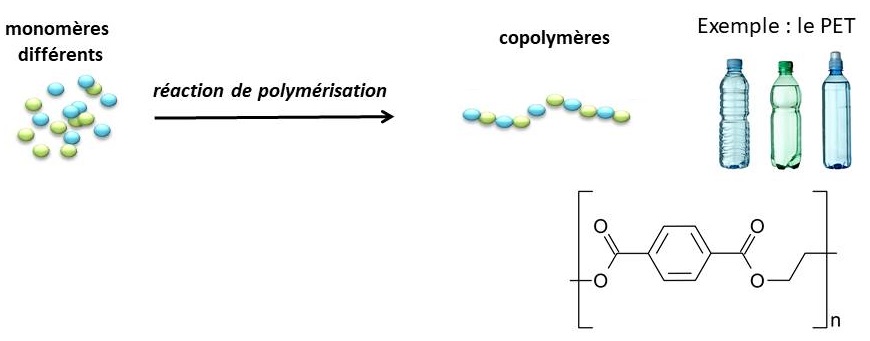
Les architectures peuvent aussi être modulées grâce à la chimie de synthèse : on parle de polymères linéaires, ramifiés, réticulés et fonctionnalisés. Ces variations apportent des propriétés physico-chimiques différentes aux matériaux comme c’est le cas par exemple avec le latex coulant de l’hévéa qui est poisseux (il contient des chaînes linéaires de polyisoprène) mais qui devient élastique après vulcanisation (réticulation des chaînes grâce à l’ajout de soufre).
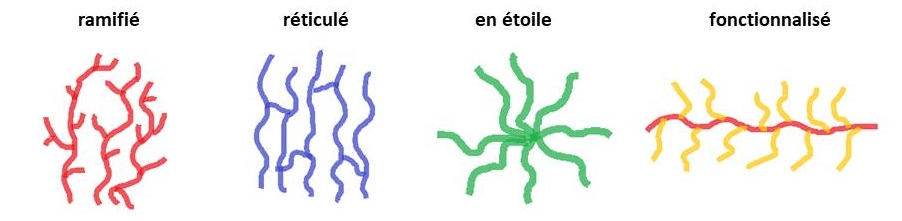
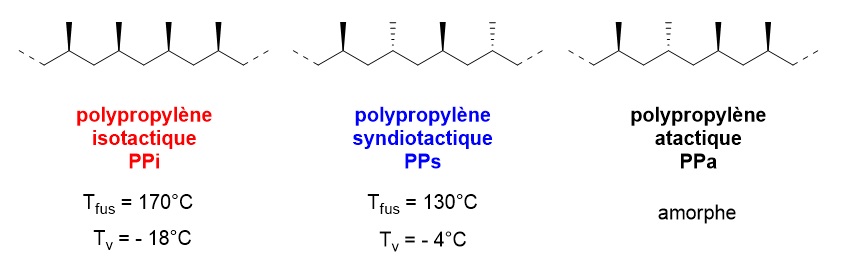
La microstructure et notamment la stéréorégularité et la régiorégularité dans l’enchaînement des monomères peuvent aussi avoir une influence sur les propriétés des matériaux. La température de fusion et la température de transition vitreuse seront par exemple différentes. On trouve de nombreuses pièces moulées en polypropylène pour la construction automobile, notamment les pare-chocs, les tableaux de bord, l’habillage de l’habitacle et les réservoirs d’essence et de liquide de frein. Le polypropylène peut être synthétisé industriellement par catalyse en présence de métallocène (on obtient un enchaînement syndiotactique des unités propyle) ou en présence de catalyseurs de Ziegler-Natta (on obtient un PP isotactique). Alors que le PPi correspond à un enchaînement de centres stéréogènes de même configuration ((R) ou (S)), le PPs correspond à une alternance de centres (R) et (S). Les températures de fusion (Tfus) et de transition vitreuse (Tv) sont différentes pour ces deux matériaux. Le polypropylène non régulier est quant à lui amorphe : il ne présente donc aucun intérêt industriel.
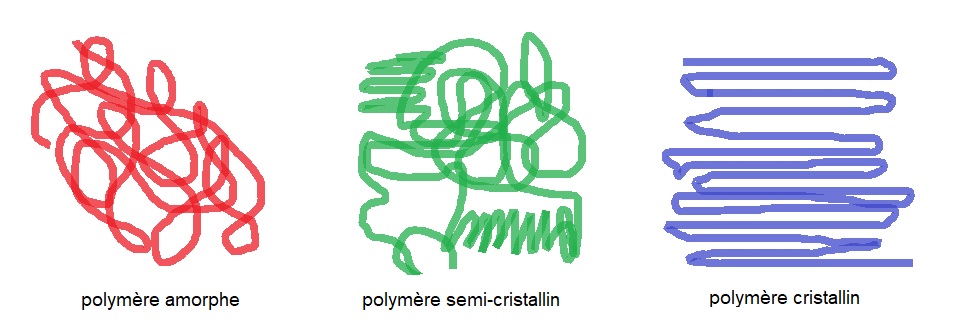
On différencie les polymères linéaires cristallins, semi-cristallins et les polymères amorphes. Les polymères semi-cristallins possèdent des cristallites, c’est-à-dire des zones dans lesquelles les chaînes de polymères s’empilent entre elles pour former des zones cristallines dans une matrice amorphe. Le polymère semi-cristallin sera caractérisé par son taux de cristallinité. La cristallinité d’un polymère semi-cristallin sera favorisé par des interactions entre les chaînes : « liaisons faibles » (liaisons physiques, par exemple interactions moléculaires de faible énergie (van der Waals) ou de moyenne énergie (liaisons hydrogène)). Une chaîne peut être en partie dans un cristallite et en partie dans une zone amorphe, voire faire partie de deux cristallites. Le passage du matériau de l’état solide à l’état liquide visqueux par chauffage s’effectue lorsque l’agitation thermique rompt ces interactions. Une élévation de température entraîne une séparation des longues chaînes moléculaires : elles glissent entre elles. Ce mécanisme est réversible (le liquide visqueux se solidifie par refroidissement car les chaînes se rapprochent). Le taux de cristallinité dépend donc également du traitement thermique subit par le polymère. Lorsqu’un polymère linéaire est chauffé puis lentement refroidi, les chaînes tendent à glisser entre elles puis à s’empiler pour former lentement des zones de cristallisation. Le taux de cristallinité sera élevé. Si le refroidissement est rapide (trempe), il ne peut pas se produire de cristallisation : le matériau sera alors vitreux.
La cristallinité d’un polymère semi-cristallin est favorisée par une microstructure régulière et ordonnée (tacticité) ayant pour conséquence une densité élevée. Ainsi le polypropylène isotactique de densité de 0,92 à 0,94 possède un taux de cristallinité entre 70 et 80% tandis que le PPs de densité 0,90 possède un taux de cristallinité de 30 à 40%. Le PP atactique, polymère amorphe avec un taux de cristallinité de 0% a une densité faible de 0,80.
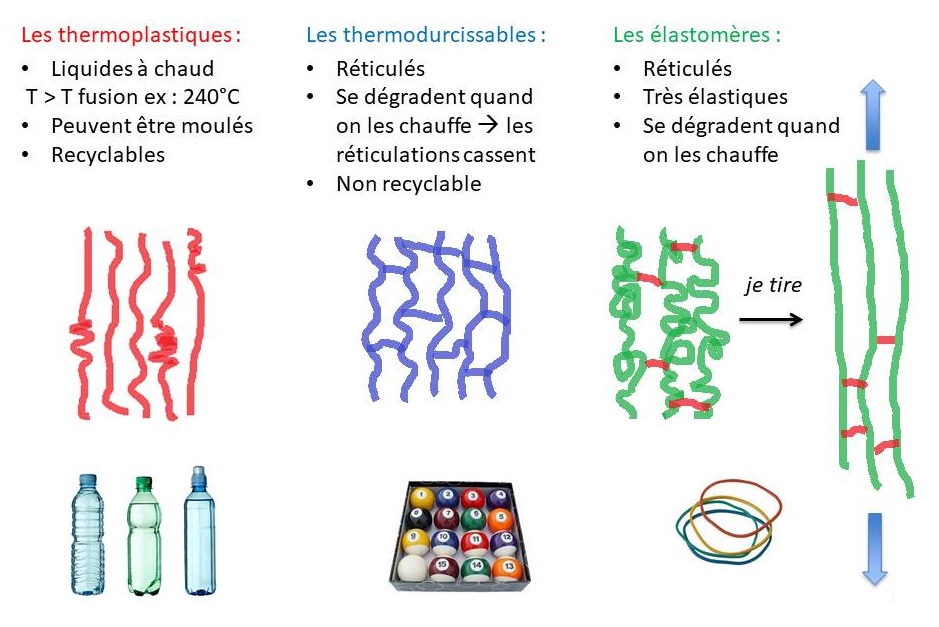
Les polymères peuvent être classés en trois grandes familles : les thermoplastiques, les élastomères et les thermodurcissables. Les thermoplastiques sont constitués de chaînes linéaires. Ils sont en général semi-cristallins, c’est-à-dire qu’ils possèdent des zones cristallines. Ils possèdent une température de fusion (Tfus) mais aussi une température de transition vitreuse (Tv). En dessous de la Tv, le matériau est cassant comme du verre. Au-dessus de la Tv, les chaînes glissent entre elles et le matériau devient mou. Lorsque le polymère est utilisé en dessous de sa Tv il est dur. Sa rigidité dépendra aussi de son taux de cristallinité. Ce type de polymère est recyclable car une fois chauffé à sa température de fusion, il peut être de nouveau mis en forme. On pourra ajouter un plastifiant pour rendre le matériau moins cassant et plus souple (des dérivés phtalates par exemple). Le polychlorure de vinyle (Tv = 80°C), le polystyrène (Tv = 100°C), le polyméthacrylate de méthyle (Tv = 108°C) ou le polycarbonate (Tv = 150°C) sont des thermoplastiques utilisés comme matériaux rigides. Le polyéthylène basse densité (PEBD) possède quant à lui une Tv aux alentours de -110°C ce qui lui confère une certaine souplesse à sa température d’utilisation. Les élastomères sont fabriqués à partir de polymères linéaires amorphes dont la force de cohésion entre les chaînes est très faible. Ils ont une température de transition vitreuse qui est faible (en général en-dessous de – 40°C) mais pas de température de fusion. Ils sont faiblement réticulés afin de leur donner des propriétés élastiques et produire des élastomères. Les thermodurcissables, enfin, sont des polymères constitués d’une forte réticulation créant un réseau 3D dense. C’est le cas par exemple de certaines mousses polyuréthanes utilisées comme joint d’étanchéité ou bien des colles résines époxy. Ils sont en général synthétisé à chaud et deviennent solides irréversiblement (réaction de polymérisation qui crée des liaisons covalentes). Chauffé une nouvelle fois, le thermodurcissable va se dégrader. Il n’est donc pas possible de le recycler.
UN CAS CONCRET
La grande famille des polyéthylènes est constituée du polyéthylène basse densité linéaire PEBDL, du polyéthylène basse densité PEBD et du polyéthylène haute densité PEHD. Chacun possède ses propres propriétés thermiques et mécaniques alors qu’ils sont issus du même monomère : l’éthylène venant de la raffinerie pétrolière. C’est la polymérisation radicalaire à très haute pression (1800 à 3000 bar) et à 200°C qui a d’abord été utilisée pour produire le polyéthylène (en 1938). Les réactions secondaires de terminaison entraînent la formation d’un matériau caractérisé par une distribution large de la taille des chaînes et la présence de ramifications plus ou moins longues irrégulièrement distribuées. Il en résulte une densité faible de 0,92 et une grande souplesse des films en PEBD (sacs plastiques de supermarché). Sa résistance mécanique est cependant plus faible que les autres types de PE. Le polyéthylène haute densité a quant à lui été synthétisé pour la première fois en 1953 par catalyse de Ziegler-Natta. Le PEHD possède une densité élevée (0,95), une distribution étroite de la taille des chaînes et pas de ramifications. Cette polyoléfine semi-cristalline est utilisée comme thermodurcissable dans la confection des canoës, des bidons d’huile, des bouteilles de lait ou de lessive. Il est flexible et rigide, translucide et très résistant aux attaques chimiques tels que les oxydants.
Alors que le PEHD est rigide, le PEBD est souple. Le polyéthylène basse densité linéaire (PEBDL), quant à lui, est en fait un copolymère fabriqué à partir d’éthylène et d’oléfines avec une double-liaison terminale : propène, butène. Il se trouve sous forme de chaînes linéaires avec des ramifications courtes et se synthétise par catalyse de Ziegler Natta. Sa densité vaut 0,92 comme le PEBD et sa flexibilité lui permet d’être utilisée comme film plastique, tout comme son cousin le PEBD. Cependant le PEBDL est beaucoup plus résistant à la rupture car sa distribution est plus étroite.
Pour en savoir plus :
article écrit par Carine Robert